- Estudio observacional con medicamentos de seguimiento prospectivo: todo estudio observacional con medicamentos en el que los sujetos son seguidos durante un periodo de tiempo hasta que acontece la variable de resultado, y esta todavía no se ha producido en el momento del inicio del estudio.
- Inicio del estudio: fecha en la que se incluye al primer paciente en el estudio o, en estudios con fuentes de información secundarias, la fecha en la que se inicia la extracción de la información.
- Finalización del estudio: fecha en la que se encuentra completamente disponible el conjunto mínimo de datos requerido para llevar a cabo el análisis estadístico que aportará los resultados relativos al objetivo primario del estudio.
- Fuente de información o fuente de los datos: origen de los datos que se utilizan para la realización del estudio. Se considerará primaria cuando la información se obtenga directamente del sujeto participante o del profesional sanitario por motivo del estudio. Se considerará secundaria cuando la información provenga de datos ya existentes, como por ejemplo la historia clínica del sujeto participante.
- Modificación sustancial: todo cambio, a partir de la obtención del dictamen favorable del Comité de Ética de la Investigación con medicamentos (en adelante, CEIm), de cualquier aspecto del estudio observacional que pueda tener repercusiones importantes en la seguridad, bienestar físico o mental de los sujetos participantes, o que pueda afectar a los resultados obtenidos en el estudio y a su interpretación, así como la inclusión de nuevas fuentes de financiación.
- Programa de apoyo a pacientes: sistema organizado en el que un titular de autorización de comercialización recibe y recoge información de sujetos individuales relacionada con la utilización de sus medicamentos.
- Promotor: individuo, empresa, institución u organización responsable de iniciar, gestionar y organizar la financiación de un estudio observacional con medicamentos.
- Investigado: persona encargada de la realización del estudio observacional con medicamentos.
- Investigador principal: investigador responsable de un equipo de investigadores que realizan un estudio observacional con medicamentos.
- Investigador coordinado: investigador responsable de la coordinación de los investigadores de los centros participantes en un estudio que se realice en más de un centro, servicio o establecimiento sanitario.
- Consentimiento informado: la expresión libre y voluntaria por parte de un sujeto participante en un estudio observacional con medicamentos, de su voluntad de participar en un estudio determinado tras haber sido informado de todos los aspectos del mismo que sean pertinentes para su decisión de participar o, en el caso de los sujetos menores o incapaces, una autorización o acuerdo de sus representantes legalmente designados para incluirlos en el estudio.
- Protocolo: documento donde se describen los objetivos, el diseño, la metodología, las consideraciones estadísticas y la organización de un estudio observacional con medicamentos. El término «protocolo» comprende las sucesivas versiones de los protocolos y sus modificaciones.
Fuente: Real Decreto 957/2020, de 3 de noviembre, por el que se regulan los estudios observacionales con medicamentos de uso humano
- Centro sanitario: el conjunto organizado de profesionales, instalaciones y medios técnicos que realiza actividades y presta servicios para cuidar la salud de los pacientes y usuarios.
- Certificado médico: la declaración escrita de un médico que da fe del estado de salud de una persona en un determinado momento.
- Consentimiento informado: la conformidad libre, voluntaria y consciente de un paciente, manifestada en el pleno uso de sus facultades después de recibir la información adecuada, para que tenga lugar una actuación que afecta a su salud.
- Documentación clínica: el soporte de cualquier tipo o clase que contiene un conjunto de datos e informaciones de carácter asistencial.
- Historia clínica: el conjunto de documentos que contienen los datos, valoraciones e informaciones de cualquier índole sobre la situación y la evolución clínica de un paciente a lo largo del proceso asistencial.
- Información clínica: todo dato, cualquiera que sea su forma, clase o tipo, que permite adquirir o ampliar conocimientos sobre el estado físico y la salud de una persona, o la forma de preservarla, cuidarla, mejorarla o recuperarla.
- Informe de alta médica: el documento emitido por el médico responsable en un centro sanitario al finalizar cada proceso asistencial de un paciente, que especifica los datos de éste, un resumen de su historial clínico, la actividad asistencial prestada, el diagnóstico y las recomendaciones terapéuticas.
- Intervención en el ámbito de la sanidad: toda actuación realizada con fines preventivos, diagnósticos, terapéuticos, rehabilitadores o de investigación.
- Libre elección: la facultad del paciente o usuario de optar, libre y voluntariamente, entre dos o más alternativas asistenciales, entre varios facultativos o entre centros asistenciales, en los términos y condiciones que establezcan los servicios de salud competentes, en cada caso.
- Médico responsable: el profesional que tiene a su cargo coordinar la información y la asistencia sanitaria del paciente o del usuario, con el carácter de interlocutor principal del mismo en todo lo referente a su atención e información durante el proceso asistencial, sin perjuicio de las obligaciones de otros profesionales que participan en las actuaciones asistenciales.
- Paciente: la persona que requiere asistencia sanitaria y está sometida a cuidados profesionales para el mantenimiento o recuperación de su salud.
- Servicio sanitario: la unidad asistencial con organización propia, dotada de los recursos técnicos y del personal cualificado para llevar a cabo actividades sanitarias.
- Usuario: la persona que utiliza los servicios sanitarios de educación y promoción de la salud, de prevención de enfermedades y de información sanitaria.
Fuente: Ley 41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente y de derechos y obligaciones en materia de información y documentación clínica
- Producto sanitario: cualquier instrumento, dispositivo, equipo, programa informático, material u otro artículo, utilizado solo o en combinación, incluidos los programas informáticos destinados por su fabricante a finalidades específicas de diagnóstico y/o terapia y que intervengan en su buen funcionamiento, destinado por el fabricante a ser utilizado en seres humanos con fines de:
- 1.º Diagnóstico, prevención, control, tratamiento o alivio de una enfermedad,
- 2.º diagnóstico, control, tratamiento, alivio o compensación de una lesión o de una deficiencia,
- 3.º investigación, sustitución o modificación de la anatomía o de un proceso fisiológico,
- 4.º regulación de la concepción,y que no ejerza la acción principal que se desee obtener en el interior o en la superficie del cuerpo humano por medios farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales medios.
- Accesorio: un artículo que, sin ser un producto sanitario, es destinado específicamente por el fabricante a ser utilizado de forma conjunta con un producto para que este último pueda utilizarse de conformidad con la finalidad prevista para el producto por su fabricante.
- Producto sanitario para diagnóstico in vitro: Cualquier producto sanitario que consista en un reactivo, producto reactivo, calibrador, material de control, estuche de instrumental y materiales, instrumento, aparato, equipo o sistema, utilizado solo o en asociación con otros, destinado por el fabricante a ser utilizado «in vitro» para el estudio de muestras procedentes del cuerpo humano, incluidas las donaciones de sangre y tejidos, sólo o principalmente con el fin de proporcionar información relativa a un estado fisiológico o patológico, o relativa a una anomalía congénita, o para determinar la seguridad y compatibilidad con receptores potenciales, o para supervisar medidas terapéuticas.
- Los recipientes para muestras se considerarán productos sanitarios para diagnóstico «in vitro». Por «recipientes para muestras» se entienden los productos, tanto si en ellos se ha hecho el vacío como si no, destinados específicamente por el fabricante a la contención directa y a la conservación de muestras procedentes del cuerpo humano para un examen diagnóstico «in vitro».
- No se considerarán productos sanitarios para diagnóstico «in vitro» los artículos de uso general en laboratorio, salvo cuando, por sus características, estén destinados específicamente por el fabricante a usarse en exámenes diagnósticos «in vitro».
- Producto a medida: un producto sanitario fabricado específicamente según la prescripción escrita de un facultativo especialista, en la que éste haga constar bajo su responsabilidad las características específicas de diseño, y que se destine únicamente a un paciente determinado.
- Los productos fabricados según métodos de fabricación continua o en serie que necesiten una adaptación para satisfacer necesidades específicas del médico o de otro usuario profesional no se considerarán productos a medida.
- Producto destinado a investigaciones clínicas: cualquier producto destinado a ser puesto a disposición de un facultativo especialista para llevar a cabo las investigaciones contempladas en el apartado 2.1 del anexo X, efectuadas en un entorno clínico humano adecuado.
- Fabricante: la persona física o jurídica responsable del diseño, fabricación, acondicionamiento y etiquetado de un producto sanitario con vistas a la puesta en el mercado de éste en su propio nombre, independientemente de que estas operaciones sean efectuadas por esta misma persona o por un tercero por cuenta de aquélla.
- Las obligaciones a que están sujetos los fabricantes en virtud de este real decreto, se aplicarán asimismo a la persona física o jurídica que monte, acondicione, trate, renueve totalmente y/o etiquete uno o varios productos prefabricados y/o les asigne una finalidad como producto con vistas a la puesta en el mercado de los mismos en su propio nombre. El presente párrafo no se aplicará a la persona que, sin ser fabricante con arreglo al párrafo primero, monte o adapte con arreglo a su finalidad prevista productos ya comercializados, para un paciente determinado.
- Finalidad prevista: la utilización a la que se destina el producto sanitario según las indicaciones proporcionadas por el fabricante en el etiquetado, las instrucciones de utilización y/o el material publicitario.
- Puesta en el mercado: la primera puesta a disposición, a título oneroso o gratuito, de un producto sanitario, no destinado a investigaciones clínicas, con vistas a su distribución y/o utilización en el mercado comunitario, independientemente de que se trate de un producto nuevo o totalmente renovado.
- Puesta en servicio: la fase en la que un producto, que está listo para ser utilizado en el mercado comunitario por primera vez con arreglo a su finalidad prevista, es puesto a disposición del usuario final.
- Representante autorizado: cualquier persona física o jurídica establecida en la Unión Europea, designada expresamente por el fabricante, que actúe en su lugar y a la que puedan dirigirse las autoridades y organismos en la Unión Europea en lugar de al fabricante por lo que respecta a las obligaciones de éste con arreglo a este real decreto.
- Datos clínicos: la información en materia de seguridad y/o prestaciones derivadas del uso de un producto; los datos clínicos se obtienen de:
- 1.º la investigación clínica del producto en cuestión, o
- 2.º la investigación clínica u otros estudios mencionados en publicaciones científicas, de un producto similar cuya equivalencia con el producto en cuestión pueda demostrarse, o
- 3.º informes publicados o no sobre otras experiencias clínicas con el producto en cuestión, o con un producto similar cuya equivalencia con aquel pueda demostrarse.
- Subcategoría de productos: conjunto de productos con áreas comunes de finalidades previstas o tecnología común.
- Grupo de productos genéricos: conjunto de productos con idénticas o similares finalidades previstas o tecnología común, lo que permite clasificarlos de forma genérica sin mencionar sus características específicas.
- Producto de un solo uso: producto destinado a ser utilizado una sola vez en un único paciente.
- Facultativo especialista: médico o cualquier otra persona que, en virtud de sus cualificaciones profesionales, se encuentra legalmente autorizado para extender la prescripción o realizar la investigación de que se trate.
- Importador: toda persona física o jurídica establecida en la Unión Europea que introduce un producto de un tercer país en territorio comunitario.
- Distribuidor: toda persona física o jurídica de la cadena de suministro distinta del fabricante o del importador que comercializa un producto.
- Comercialización: todo suministro, remunerado o gratuito, para su distribución o utilización en el mercado comunitario en el transcurso de una actividad comercial.
- Promotor: fabricante, representante autorizado u otro individuo u organización que asume la responsabilidad de la iniciación y/o puesta en práctica de una investigación clínica.
Fuente: Real Decreto 1591/2009, de 16 de octubre por el que se regulan los productos sanitarios
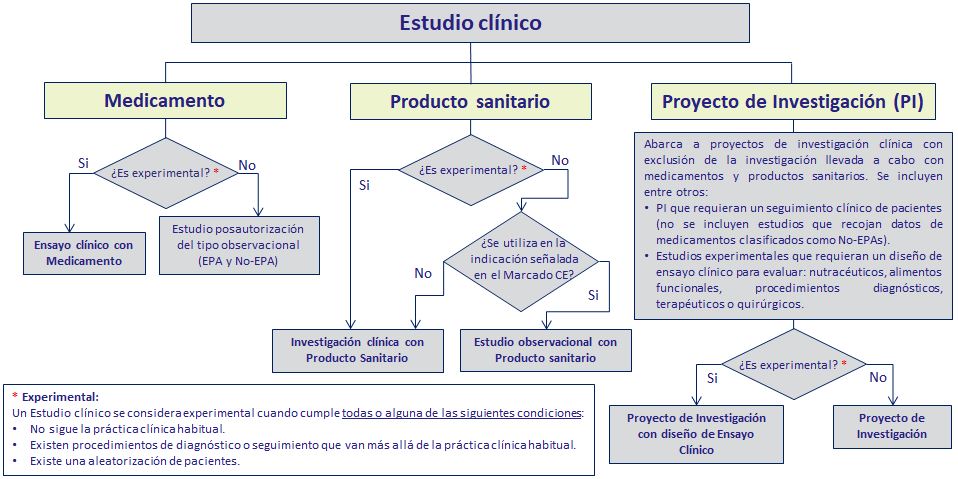
- Ensayo clínico (EECC): Es un estudio analítico y experimental, con direccionalidad anterógrada (de la exposición al efecto) y temporalidad concurrente (el investigador está presente en el momento de la exposición y en el del efecto).
- Ensayo clínico de baja intervención: Es un estudio analítico y experimental, en el que el medicamento ya está autorizado. Los procedimientos complementarios entrañan riesgo o carga adicional mínimo comparado con la práctica clínica habitual
- Estudio Postautorizado de tipo observacional (EPA): "Cualquier estudio clínico o epidemiológico realizado durante la comercialización de un medicamento según las condiciones autorizadas en su ficha técnica, o bien en condiciones normales de uso, en el que el medicamento o los medicamentos de interés son el factor de exposición fundamental investigado. Este estudio podrá adoptar la forma de un ensayo clínico o de un estudio observacional".
EPA-AS: Aquellos estudios observacionales de seguimiento prospectivo, promovidos por Autoridades Sanitarias o financiados con fondos públicos, para los que se establece un procedimiento simplificado para suautorización.
EPA-LA: Aquellos estudios que sean una condición establecida en el momento de la autorizaciónde un medicamento, o bien constituya unaexigencia de la autoridad competente para aclarar cuestiones relativas a la seguridad delmedicamento, o forme parte del plan de gestión de riesgos.
EPA-SP: Estudio postautorizacion de tipo observacional de seguimiento prospectivo. Estudio en el que los pacientes se seleccionan por su exposición y se siguen durante el tiempo suficiente, siendo el periodo de estudio posterior al inicio de la investigación. Se excluyen los estudios pertenecientes a las categorías anteriores. Se incluyen estudios con o sin grupo control. También incluye estudios cuyo seguimiento sea ambispectivo. Estos estudios, por su diseño, son susceptibles de ser utilizados como instrumento para la inducción a la prescripción.
EPA-OD: Aquellos estudios cuyo diseño no corresponde a EPA observacional de seguimiento prospectivo. Serán aquellos estudios transversales o cuyo seguimiento sea retrospectivo (casos y controles, cohorte retrospectivo, etc,...).
EPA-PS: Investigaciones clínicas realizadas con productos sanitarios que ostentan el marcado CE en condiciones normales de utilizacion,, las prestaciones de los productos corresponden a las atribuidas por el fabriante
NO EPA: Aquellos estudios en los que el medicamento no es el factor de exposición fundamental investigado. Estudios epidemiológicos (de incidencia o prevalencia) de enfermedades.